
White blood cells, or leukocytes, are the body’s first and second lines of defense against foreign organisms and particles. However, few drugs target these cells’ production and movement for clinically useful purposes. A new study published in the journal Immunity explores the signaling molecule landscape to identify potentially druggable targets for leukocyte migration into the bloodstream.
 Study: Small-molecule CBP/p300 histone acetyltransferase inhibition mobilizes leukocytes from the bone marrow via the endocrine stress response. Image Credit: Rost9 / Shutterstock
Study: Small-molecule CBP/p300 histone acetyltransferase inhibition mobilizes leukocytes from the bone marrow via the endocrine stress response. Image Credit: Rost9 / Shutterstock
Leukocytes, including neutrophils, monocytes, and B lymphocytes, are formed in the bone marrow from blood-forming precursor cells and in a few other specialized organs. They are held in the bone marrow until they are released into the circulation.
There are two leukocyte compartments in the blood and peripheral tissues, which show changes in size with varying bodily states. For instance, when the body is injured, stressed, or infected, the number of leukocytes in the affected tissue alters and returns to normal once the threat is contained.
Multiple regulatory steps take part in leukocyte breakdown as well as movement to different sites where they are needed. These originate in the central nervous system (CNS) in response to peripheral signals, being regulated by neural circuits in which both the sympathetic nervous system and the hypothalamo-pituitary-adrenal (HPA) axis participate.
These signals work to increase bone marrow hemopoiesis, recruit leukocytes into the blood and other tissues where they are required, and ensure they return to normal levels once the challenge has been surmounted.
In some disease conditions, this homeostatic control is lost, thus leading to abnormal counts, such as bone marrow failure on the one hand or acute leukemia on the other. As yet, though, few medications can help correct such dysregulation by modifying the rate of production, breakdown, or migration of leukocytes, whether in blood cancer, chronic inflammation, or acute hyperinflammatory states.
Among available medications are the granulocyte colony-stimulating factor (G-CSF) family, CXC-motif chemokine receptor 4 (CXCR4) antagonists such as plerixafor/AMD3100), or inhibitors of the integrin very late antigen 4 (VLA4). G-CSF is, for instance, used to correct neutropenia in patients on chemotherapy but is less useful in patients with acute febrile conditions involving low neutrophil counts. Moreover, G-CSF can cause adverse effects in some patients.
The need to know more about this field of pharmacology motivated the current study. It focuses on a small molecule called E1A-associated protein p300 (EP300 or p300), which is seen to be newly acquired during the leukemic phase of a condition called severe congenital neutropenia (SCN).
The loss of function of this gene has led to reduced blood cell production if deleted before birth but high or leukemic leukocyte counts in later life. This has an ortholog, cyclic-adenosine-monophosphate-response-element-binding protein (CREBBP, also known as “CBP,” with 90% homology of sequence. One of the 8 domains in this gene is responsible for histone acetyltransferase (HAT) activity and contains a mutation in SCN that causes leukemic transformation.
In this case, this domain might be druggable to produce “leukocytosis on demand” by altering the sizes of different leukocyte compartments.
What did the study show?
The scientists found that inhibiting the CBP/p300 domain with its HAT activity by the small molecule inhibitor A485 led to a reversible competitive inhibition of HAT enzyme activity, especially for CBP and p300 compared to other HATs. As expected, this led to a rapid rise in the levels of acetyl CoA within bone marrow macrophages in mouse models. The result was rapid leukocytosis.
This was found to be a dose-dependent action and did not wane with repeated administration. When another type of CBP/p300 HAT inhibitor (C646) was used, the same effect was observed, confirming the mechanism of action. Conversely, inhibitors of DNA binding by the protein or of another HAT found in mammals failed to cause leukocytosis.
A485 levels in the blood rapidly rose when injected into the mice, accumulating in bone marrow, adipose tissues, liver, spleen, and kidney, but not the brain. Leukocyte counts rose in parallel, including neutrophils, lymphocytes, and monocytes. A week later, no evidence of drug administration was observable, suggesting a transient effect.
The rise in leukocyte counts was comparable with that achieved by G-CSF, though somewhat faster for neutrophils. When both were given, significantly higher neutrophil counts resulted. However, after 24 hours, all three blood cell types were raised with G-CSF vs A485.
This indicates a shorter and different action of A485 compared to G-CSF.
To extend the observations to human subjects, the researchers looked at data from a cohort of patients with a rare disease called Rubinstein-Taybi syndrome (RSTS), where CREBBP and EP300 mutations occur. About two-thirds had high leukocyte counts, with 70% showing mutations in the HAT domain. As expected, this group was more likely to show leukocytosis than the other group, where HAT was spared.
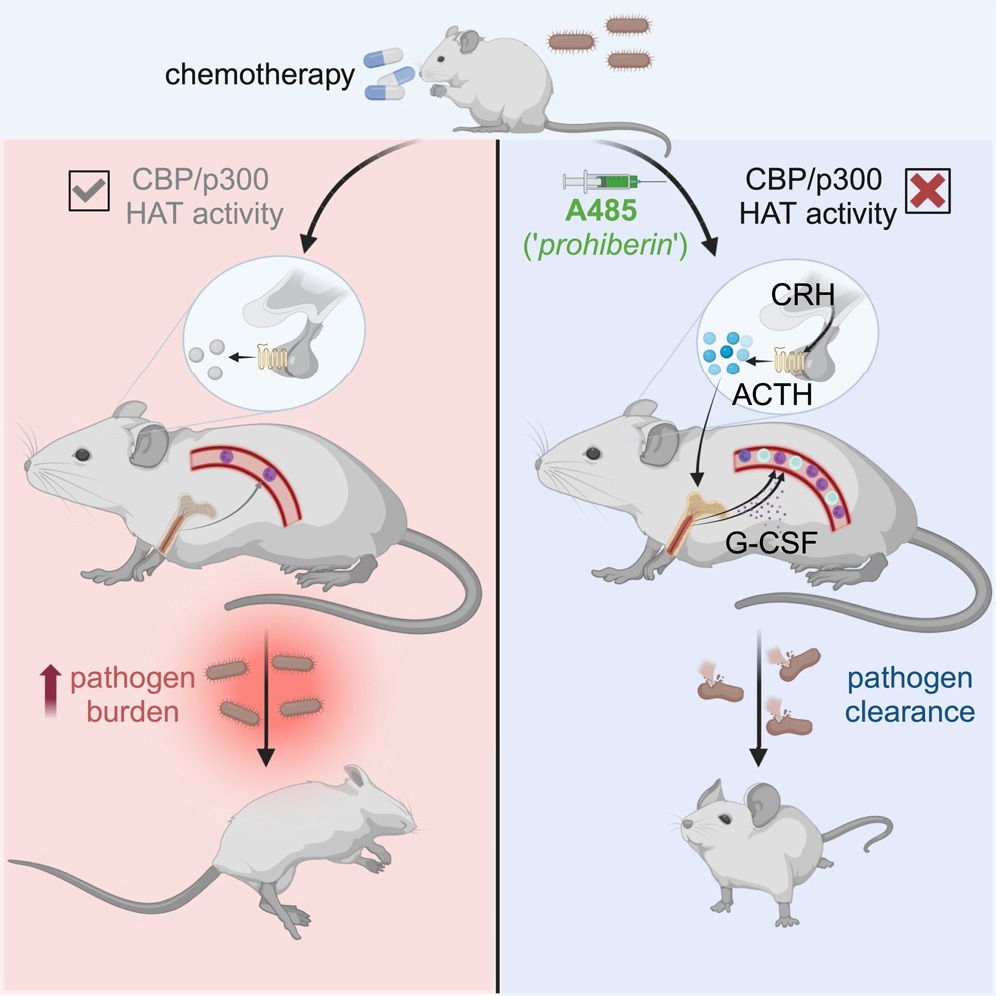
Does this observation have clinical utility? To find out, they tested the effect of A485 in a cohort of mice with myelodysplastic syndrome (MDS), finding that the small molecule kept the leukocyte count normal. Secondly, they induced severe neutropenia by a course of chemotherapy in a mouse model, showing that A485 led to acute recovery of leukocyte counts.
Then, they introduced the organism Listeria monocytogenes in a sepsis-inducing dose in mice with chemotherapy-induced pancytopenia. Neutrophils are vital to the immune defense against this microbe. After infection set in, they injected A485 vs. vehicle in controls.
While those treated with the vehicle became sick and died of sepsis, A485 in a single dose led to improved survival, with fewer bacteria being recovered from treated animals. A485 mobilizes leukocytes from the bone marrow, which is the mechanism of leukocytosis. In contrast, there was no emergency hematopoiesis in the bone marrow.
Different subsets of leukocytes responded to distinct pathways triggered by A485. These involve both G-CSF-dependent and –independent pathways of neutrophilia, but other pathways for lymphocytosis.
Moreover, A485 uses neurohumoral pathways, specifically the HPA axis, to induce leukocytosis, as seen by the increased levels of glucocorticoids in the blood after A485 administration. The leukocytosis response triggered by the HPA activation does not rely on glucocorticoids, however, but occurs in response to CRHR1-regulated signals, including the adrenocorticotropic hormone (ACTH), that occurs with the loss of HPA feedback signals.
While neutrophils increase with ACTH administration, lymphocyte counts increase only with glucocorticoid blockade, indicating that both are regulated differently.
What are the implications?
“Competitive, reversible, small-molecule-mediated inhibition of the CBP/p300 HAT domain triggers acute and transient leukocyte mobilization from the bone marrow.” Further research is required to identify which clinical contexts are ideal for this drug. A485 may be better if only a rapid short increase in neutrophils is required, while long-term recovery of blood cell production in the bone marrow may call for G-CSF.
The timing of administration for good results also needs to be defined since patients with neutropenic sepsis present at various time points and stages. Moreover, the value of such drugs in bacterial or viral, rather than listerial, sepsis remains unexplored.
However, as reported by earlier researchers, it has anti-tumor effects, which could make it valuable in adjuvant therapy for cancer patients. The current study also sheds light on the role of ACTH, rather than its downstream products, glucocorticoids, on leukocyte homeostasis and G-CSF activity.






